
KAPRUVIA 50 microgrammes-mL, solution injectable, boîte de 12 flacons (verre) de 1 mL
Retiré du marché le : 16/12/2022
Dernière révision : 23/06/2022
Taux de TVA : 2.1%
Laboratoire exploitant : VIFOR FRANCE
Source :

Kapruvia est indiqué dans le traitement du prurit sévère associé à la maladie rénale chronique chez les patients adultes sous hémodialyse (voir rubrique Propriétés pharmacodynamiques).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Hyperkaliémie
Une hyperkaliémie survient fréquemment chez les patients atteints de maladie rénale chronique sous hémodialyse. Dans les études cliniques contrôlées par placebo, un taux numériquement plus élevé d'événements indésirables d'hyperkaliémie a été rapporté chez les patients traités par difélikéfaline (4,7 % ; 20/424 patients) par rapport au placebo (3,5 % ; 15/424 patients). Aucun lien de causalité n'a été établi. Une surveillance fréquente des taux de potassium est recommandée.
Insuffisance cardiaque et fibrillation auriculaire
La difélikéfaline n'a pas été étudiée chez les patients présentant une insuffisance cardiaque de classe New York Heart Association IV. Dans les études cliniques pivots, un léger déséquilibre numérique des événements d'insuffisance cardiaque et de fibrillation auriculaire a été observé chez les patients traités par difélikéfaline par rapport au placebo, en particulier chez les patients ayant des antécédents médicaux de fibrillation auriculaire qui ont interrompu ou qui n'ont pas pris leur traitement contre la fibrillation auriculaire. Aucun lien de causalité n'a été établi.
Patients présentant une altération de la barrière hémato-encéphalique
La difélikéfaline est un agoniste spécifique des récepteurs opioïdes kappa périphériques sans passage dans le système nerveux central (SNC). L'intégrité de la barrière hémato-encéphalique (BHE) est importante pour réduire l'absorption de difélikéfaline dans le SNC (voir rubrique Propriétés pharmacodynamiques). Les patients présentant des anomalies cliniquement importantes de la BHE (par exemple, des tumeurs cérébrales malignes primaires, des métastases du SNC ou d'autres affections inflammatoires, une sclérose en plaques active, une maladie d'Alzheimer avancée) peuvent présenter un risque de pénétration de la difélikéfaline dans le SNC. Kapruvia doit être prescrit avec prudence chez ces patients en tenant compte du rapport bénéfice/risque de chacun et en surveillant les éventuels effets sur le SNC.
Sensation de vertige et somnolence
Des
sensations de vertige et de la somnolence sont survenues chez des
patients prenant de la difélikéfaline et peuvent s'atténuer au cours du
temps avec la poursuite du traitement
(voir rubrique Effets indésirables). L'utilisation
concomitante d'antihistaminiques sédatifs, d'analgésiques opioïdes et
d'autres dépresseurs du SNC et peut augmenter la probabilité de ces
effets indésirables. Ces médicaments doivent être utilisés avec
prudence pendant le traitement par la difélikéfaline (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Par rapport au placebo, l'incidence de la somnolence a été plus élevée
chez les sujets âgés de 65 ans et plus traités par la difélikéfaline
(7,0 %) que chez les sujets âgés de moins de 65 ans traités par la
difélikéfaline (2,8 %).
Excipients à effet notoire
Ce médicament contient moins de 1 mmol de sodium par flacon, c.-à-d. qu'il est essentiellement sans sodium.
Résumé du profil de sécurité
Dans les études cliniques de phase 3 contrôlées et non contrôlées par placebo, environ 6,6 % des patients ont présenté au moins un effet indésirable pendant le traitement par difélikéfaline. Les effets indésirables les plus fréquents étaient la somnolence (1,1 %), les sensations de vertige (0,9 %), la paresthésie (y compris l'hypoesthésie, la paresthésie buccale et l'hypoesthésie buccale) (1,1 %), les céphalées (0,6 %), les nausées (0,7 %), les vomissements (0,7 %), la diarrhée (0,2 %) et les modifications de l'état mental (y compris l'état confusionnel) (0,3 %). La plupart de ces événements étaient d'une sévérité légère ou modérée, n'ont pas entraîné de conséquences délétères et se sont résolus avec la poursuite du traitement. Aucun événement n'était grave et l'incidence des événements conduisant à un arrêt du traitement était ≤ 0,5 % pour tous les effets indésirables mentionnés ci- dessus.
Liste tabulée des effets indésirables
Les effets indésirables observés dans les études cliniques de phase 3 contrôlées et non contrôlées par placebo chez les patients (N = 1 306) traités par difélikéfaline sont listés dans le Tableau 1 par classe de système d'organe MedDRA et par fréquence.
Les catégories de fréquence sont définies comme suit : fréquent (≥ 1/100 à < 1/10) et peu fréquent (≥ 1/1 000 à < 1/100).
Pour chaque catégorie de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 1: Effets indésirables attribués au traitement par la difélikéfaline chez les patients hémodialysés
| Classe de système d'organe MedDRA | Fréquent | Peu fréquent |
| Affections psychiatriques | | Modifications de l'état mental1 |
| Affections du système nerveux | Somnolence, paresthésie2 | Sensations de vertige ; céphalées |
| Affections gastro-intestinales | | Nausées ; diarrhée |
2 La paresthésie inclut les termes MedDRA (‘preferred terms') de paresthésie, d'hypoesthésie, de paresthésie buccale et d'hypoesthésie buccale.
Description de certains effets indésirables
Somnolence
Une
somnolence a été rapportée comme événement indésirable sous traitement
chez 2,2 % des sujets du groupe difélikéfaline. La grande majorité de
ces événements était de sévérité légère ou modérée. Chez 0,3 % des
patients, la somnolence a conduit à un arrêt du traitement par
difélikéfaline. La somnolence a été rapportée comme événement
indésirable grave chez < 0,1 % des sujets traités par
difélikéfaline. Chez 1,1 % des patients, un lien de causalité entre la
somnolence et le traitement par difélikéfaline a été rapporté. La
somnolence est survenue au cours des 3 premières semaines de traitement
et a eu tendance à s'atténuer avec la poursuite du traitement.
Le risque de somnolence peut augmenter lors de l'utilisation
concomitante de difélikéfaline avec d'autres médicaments (voir
rubriques Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions).
Sensations de vertige
Des
sensations de vertige ont été rapportées comme événement indésirable
apparu sous traitement chez 7,9 % des sujets du groupe difélikéfaline.
La grande majorité de ces événements était de sévérité légère ou
modérée. Chez 0,5 % des patients, les sensations de vertige ont conduit
à l'arrêt du traitement par difélikéfaline. Les sensations de vertige
ont été rapportées comme événement indésirable grave chez 0,5 % des
sujets traités par difélikéfaline. Chez 0,9 % des patients, un lien de
causalité entre les sensations de vertige et le traitement par
difélikéfaline a été rapporté. Les sensations de vertige sont survenues
au cours des 9 premières semaines de traitement et ont été généralement
transitoires.
Ce risque peut augmenter lors de l'utilisation concomitante de difélikéfaline avec d'autres médicaments (voir rubriques Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions).
Modifications de l'état mental
Des
modifications de l'état mental (y compris l'état confusionnel) ont été
rapportées comme événement indésirable apparu sous traitement chez 4,4
% des sujets du groupe difélikéfaline.
La majorité de ces événements était de sévérité légère ou modérée. Chez
moins de 0,2 % des patients, les modifications de l'état mental ont
conduit à l'arrêt du traitement par difélikéfaline.
Les modifications de l'état mental ont été rapportées comme événement
indésirable grave chez 2,2 % des sujets traités par difélikéfaline.
Chez 0,3 % des patients, un lien de causalité entre les modifications
de l'état mental et le traitement par difélikéfaline a été rapporté.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté selon les modalités définies dans le Protocole d'utilisation thérapeutique et de recueil de données (voir PUT RD).
SURVEILLANCE du traitement :
- kaliémie.
NE PAS ADMINISTRER plus de 4 doses par semaine même si le nombre de séances
d'hémodialyse dans une semaine est supérieur à 4.
Grossesse
Il n'existe pas de données ou il existe des données limitées sur l'utilisation de la difélikéfaline chez la femme enceinte.
Les études effectuées chez l'animal n'ont pas mis en évidence d'effets
délétères directs ou indirects sur la reproduction (voir rubrique Données de sécurité préclinique).
Par mesure de précaution, il est préférable d'éviter l'utilisation de Kapruvia pendant la grossesse.
Allaitement
On ne sait pas si la difélikéfaline est excrétée dans le lait maternel.
Un risque pour les nouveau-nés/nourrissons ne peut être exclu.
Une décision doit être prise soit d'interrompre l'allaitement soit
d'interrompre/de s'abstenir du traitement par Kapruvia en prenant en
compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice
du traitement pour la mère.
Les études effectuées chez l'animal ont montré une excrétion de la difélikéfaline dans le lait maternel.
Fertilité
Il n'existe aucune donnée disponible sur les effets de la difélikéfaline sur la fertilité humaine. Dans les études menées chez le rat avec la difélikéfaline, aucun effet sur la fertilité n'a été observé (voir rubrique Données de sécurité préclinique).
Aucune étude d'interaction clinique n'a
été réalisée. La difélikéfaline n'est ni un inhibiteur ni un inducteur
ni un substrat des enzymes CYP450. Elle n'est pas non plus un
inhibiteur des enzymes de glucuronidation. La difélikéfaline n'est pas
un substrat ou un inhibiteur des protéines de transport (voir rubrique Propriétés pharmacocinétiques). Par conséquent, les interactions de la difélikéfaline avec d'autres médicaments sont peu probables.
L'administration concomitante de médicaments, tels que des antihistaminiques sédatifs, des analgésiques opioïdes et d'autres inhibiteurs du SNC (par exemple, clonidine, ondansétron, gabapentine, prégabaline, zolpidem, alprazolam, sertraline, trazodone) peut augmenter la probabilité de sensation de vertige et de somnolence (voir rubrique Mises en garde spéciales et précautions d'emploi).
L'utilisation de Kapruvia doit se limiter exclusivement aux centres d'hémodialyses.
Kapruvia doit être utilisé par des professionnels de santé expérimentés dans le diagnostic et le traitement des pathologies pour lesquelles la difélikéfaline est indiquée. Les autres causes de prurit - autres que celui associé à la maladie rénale chronique - doivent être exclues avant d'instaurer le traitement par difélikéfaline.
Posologie
La difélikéfaline est administrée 3 fois par semaine par injection intraveineuse en bolus dans la ligne veineuse du circuit de dialyse à la fin de la séance d'hémodialyse pendant ou après le rinçage.
La dose recommandée de difélikéfaline est de 0,5 microgramme/kg de poids sec (c'est-à-dire le poids cible après dialyse). Le volume total de la dose (mL) nécessaire à prélever doit être calculé comme suit : 0,01 × poids corporel sec (kg), arrondi au dixième le plus proche (0,1 mL). Pour les patients dont le poids sec est supérieur ou égal à 195 kg, la dose recommandée est de 100 microgrammes (2 mL). Les volumes d'injection sont détaillés dans le tableau ci-dessous :
|
Intervalle de poids (poids sec en kg) |
Volume d'injection1 (mL) |
| 40 à 44 | 0,4 |
| 45 à 54 | 0,5 |
| 55 à 64 | 0,6 |
| 65 à 74 | 0,7 |
| 75 à 84 | 0,8 |
| 85 à 94 | 0,9 |
| 95 à 104 | 1,0 |
| 105 à 114 | 1,1 |
| 115 à 124 | 1,2 |
| 125 à 134 | 1,3 |
| 135 à 144 | 1,4 |
| 145 à 154 | 1,5 |
| 155 à 164 | 1,6 |
| 165 à 174 | 1,7 |
| 175 à 184 | 1,8 |
| 185 à 194 | 1,9 |
| ≥ 195 | 2,0 |
1 Plus d'un flacon peut être nécessaire si un volume d'injection de plus de 1 mL est requis.
Un effet de la difélikéfaline sur la réduction du prurit est attendu après 2 à 3 semaines de traitement.
Omissions de prise
Si une séance d'hémodialyse n'est pas réalisée, Kapruvia doit être administré lors de la séance suivante à la même dose.
Traitement supplémentaire
Si une 4e séance d'hémodialyse doit être effectuée dans la semaine, Kapruvia doit être administré à la fin de la séance à la dose recommandée. Il ne faut pas administrer plus de 4 doses par semaine même si le nombre de séances d'hémodialyse dans une semaine est supérieur à 4. Il est peu probable qu'une 4e dose de Kapruvia entraîne une accumulation de difélikéfaline qui représenterait un problème pour la tolérance, puisque la majorité de la difélikéfaline résiduelle du traitement précédent sera éliminée par hémodialyse (voir rubriques Surdosage et Propriétés pharmacocinétiques). Toutefois, la sécurité et l'efficacité d'une 4e dose n'ont pas été complètement établies en raison de l'insuffisance de données.
Patients dont le traitement par hémodialyse est incomplet
Pour
les séances d'hémodialyse de moins d'une heure, l'administration de difélikéfaline doit être suspendue jusqu'à la séance
d'hémodialyse suivante.
Après
l'administration de la difélikéfaline chez les
patients hémodialysés, jusqu'à 70 % de difélikéfaline
est éliminé de l'organisme avant la séance d'hémodialyse suivante (voir
rubriques Surdosage et Propriétés pharmacocinétiques). Le taux
plasmatique de difélikéfaline restant au moment de
l'hémodialyse suivante est réduit d'environ 40 à 50 % dans l'heure qui suit
l'hémodialyse.
Patients présentant une insuffisance hépatique
Aucun ajustement de la dose n'est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée (voir rubrique Propriétés pharmacocinétiques). La difélikéfaline n'a pas été étudiée chez les patients présentant une insuffisance hépatique sévère (Groupe de travail sur les défaillances d'organes [ODWG, Organ DysfunctionWorking Group] du National Cancer Institute [NCI]) et son utilisation n'est donc pas recommandée dans cette population de patients.
Population âgée (≥ 65 ans)
Les recommandations posologiques pour les patients âgés sont les mêmes que pour les patients adultes.
Population pédiatrique
La
sécurité et l'efficacité de la difélikéfaline chez
les enfants âgés de 0 à 17 ans n'ont pas encore été établies.
Aucune
donnée n'est disponible.
Mode d'administration
Kapruvia ne doit pas être dilué et ne doit pas être mélangé avec d'autres médicaments.
La
difélikéfaline est éliminée par la membrane du
dialyseur et doit être administrée une fois que le sang ne circule plus dans le
dialyseur. La difélikéfaline est administrée 3 fois
par semaine par injection intraveineuse en bolus dans la ligne veineuse du
circuit de dialyse à la fin du traitement d'hémodialyse pendant ou après le
rinçage.
Lorsqu'elle
est administrée après le rinçage, au moins 10 mL de
solution injectable de chlorure de sodium à 9 mg/mL
(0,9 %) doivent être administrés après l'injection de Kapruvia.
Si la dose est administrée
pendant le rinçage, aucune solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) supplémentaire n'est nécessaire pour rincer la
ligne.
Durée de conservation :
2 ans
Précautions particulières de conservation :Ce médicament ne nécessite pas de précautions particulières de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Des
doses uniques de difélikéfaline jusqu'à 12 fois et des
doses multiples de difélikéfaline jusqu'à 5
fois la dose clinique de 0,5 microgramme/kg ont été administrées dans
des études cliniques chez des patients sous hémodialyse. Une
augmentation dose-dépendante des effets indésirables, notamment des
sensations de vertige, de la somnolence, des modifications de l'état
mental, des paresthésies, de la fatigue, de l'hypertension et des
vomissements, a été observée.
En cas de surdosage, les soins médicaux appropriés en fonction de
l'état clinique du patient doivent être dispensés. Une hémodialyse
pendant 4 heures à l'aide d'un dialyseur à haut débit a éliminé
efficacement environ 70 à 80 % de la difélikéfaline du plasma, et la
difélikéfaline n'était pas détectable dans le plasma à la fin du
deuxième des deux cycles de dialyse (voir rubrique Propriétés pharmacocinétiques).
Classe pharmacothérapeutique : tous les autres produits thérapeutiques, tous autres médicaments, code ATC : V03AX04
Mécanisme d'action
La difélikéfaline est un agoniste spécifique des récepteurs opioïdes kappa à faible pénétration dans le système nerveux central.
Les propriétés physico-chimiques de la difélikéfaline (peptide
synthétique hydrophile à base d'acide aminé D, avec une surface polaire
et une charge élevées au pH physiologique) minimisent sa
diffusion passive (perméabilité) et son transport actif à travers les
membranes, limitant ainsi sa pénétration dans le système nerveux
central.
On pense que la physiopathologie du prurit associé à la maladie rénale
chronique est multifactorielle et comprend une inflammation systémique
et un déséquilibre du système opioïde endogène (par exemple,
surexpression des récepteurs opioïdes mu et dérèglement concomitant des
récepteurs opioïdes kappa). Les récepteurs opioïdes sont connus pour
moduler les signaux de démangeaison et l'inflammation, l'activation des
récepteurs opioïdes kappa réduisant la démangeaison et produisant des
effets immunomodulateurs.
L'activation des récepteurs opioïdes kappa présents sur les neurones
sensoriels périphériques et les cellules immunitaires par la
difélikéfaline est considérée comme mécaniquement responsable des
effets antiprurigineux et anti-inflammatoires.
Efficacité et sécurité clinique
Études contrôlées par placebo
Dans deux études cliniques pivotales de
phase 3 de conception similaire, en double aveugle, randomisées et
contrôlées par placebo (KALM-1 et KALM-2), des patients atteints de
maladie rénale chronique sous hémodialyse et présentant un prurit
modéré à sévère ont reçu soit un placebo, soit 0,5 microgramme/kg
de difélikéfaline par voie intraveineuse 3 fois par semaine après
l'hémodialyse pendant 12 semaines. Un maximum de 4 doses par semaine
était autorisé chez les patients recevant une dialyse supplémentaire au
cours d'une semaine donnée. Le critère principal d'évaluation dans les
deux études était le pourcentage de patients ayant obtenu une réduction
d'au moins 3 points par rapport à l'inclusion
sur l'échelle d'évaluation
numérique des démangeaisons
(WI-NRS) à 12 semaines. Les principaux critères secondaires
d'évaluation des deux études étaient les pourcentages de patients
présentant une amélioration du score WI-NRS d'au moins 4
points après 12 semaines et les modifications de la sévérité des
démangeaisons et de la qualité de vie (QdV) liée aux démangeaisons,
mesurées par le Skindex-10 total et l'échelle de démangeaison 5-D.
Une analyse des répondeurs basée sur l'impression globale de
changement du patient a également été incluse.
Au total, 851 patients présentant un prurit modéré à sévère (WI-NRS
> 4 à l'inclusion) ont été recrutés dans les études pivots. L'âge
moyen était de 59 ans, 33,1 % étaient âgés de 65 ans et plus et 11,1 %
étaient âgés de 75 ans et plus ; 60 % des patients étaient des hommes.
Les scores WI-NRS moyens à l'inclusion étaient de 7,18 dans les deux
groupes, difélikéfaline et placebo ; les scores WI-NRS médians à
l'inclusion étaient de 7,13 (intervalle de 4,2 à 10) dans le groupe
difélikéfaline et de 7,13 (intervalle de 4,1 à 10) dans le groupe
placebo. Les autres caractéristiques de la maladie à l'inclusion
étaient comparables dans les groupes difélikéfaline et placebo : temps
écoulé depuis le diagnostic de la maladie rénale chronique (8,22 ans
contre 8,54 ans), durée du prurit (3,20 ans contre 3,31 ans) et
utilisation de médicaments destinés à soulager le prurit, tels que des
antihistaminiques, des corticoïdes, de la gabapentine ou de la
prégabaline (37,5 % contre 38 %). Dans l'ensemble des études, la
difélikéfaline a réduit de manière significative l'intensité des
démangeaisons et a amélioré la qualité de vie liée aux démangeaisons
sur 12 semaines, comme le montre le tableau 2.
Tableau 2: Résumé des critères principaux et secondaires clés dans KALM-1 et KALM-2 à la semaine 12
| | KALM-1 (n = 378) | KALM-2 (n = 473) | |||||
| Critère d'évaluation à la fin de la semaine 12 | difélikéfaline (n = 189) | Placebo (n = 189) | difélikéfaline (n = 237) | Placebo (n = 236) | |||
| Critère d'évaluation | principal | | | | | ||
| WI-NRS | | | | | |||
| Patients | avec | une | 51,0 % | 27,6 % | 54,0 % | 42,2 % | |
| amélioration ≥ 3 points | (p < 0,001) | | (p = 0,02) | | |||
| (%) | | | | | |||
| WI-NRS | | | | | |||
| Patients | avec | une | 38,9 % | 18,0 % | 41,2 % | 28,4 % | |
| amélioration ≥ 4 points | (p < 0,001) | | (p = 0,01) | | |||
| (%) | | | | | |||
| Skindex-10 | | | | | |||
| Changement par rapport | -17,2 | -12,0 | -16,6 | -14,8 | |||
| à l'inclusion | (p < 0,001) | | (p = 0,171) | | |||
| [score total] | | | | | |||
| Démangeaison 5-D | | | | | |||
| Changement par rapport | -5,0 | -3,7 | -4,9 | -3,8 | |||
| à l'inclusion | (p < 0,001) | | non applicable1 | | |||
| [score total] | | | | | |||
La figure 1 montre le pourcentage hebdomadaire moyen des patients des études KALM-1 et KALM-2 qui présentent une amélioration du score WI-NRS ≥ 3 points par rapport à l'inclusion. D'après les rapports de cotes (‘Odds ratio'), des améliorations importantes sur le plan statistique en faveur du groupe difélikéfaline ont été observées dès la semaine 3 dans l'étude KALM-1 et dès la semaine 2 dans l'étude KALM-2 et se sont poursuivies chaque semaine, jusqu'à la semaine 12 dans les deux études.
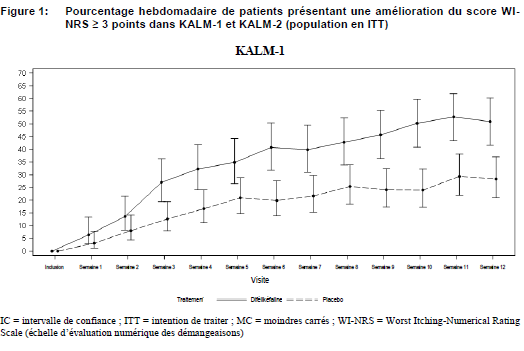
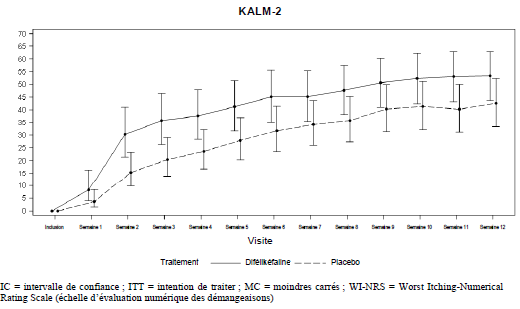
Études d'extension en ouvert
L'effet
du traitement par difélikéfaline pendant une durée allant jusqu'à 52
semaines a été évalué à l'aide de l'échelle de démangeaison 5-D dans
des phases d'extensions ouvertes à un seul bras des études KALM-1 et
KALM-2 incluant 712 patients.
Chez les patients passant du placebo à la difélikéfaline à la fin de la
phase en double aveugle, une amélioration du score de l'échelle de
démangeaison 5-D a été observée après 4 semaines de traitement, avec
une moyenne des moindres carrés du changement par rapport aux valeurs à
l'inclusion comparable à celle des patients recevant la difélikéfaline
depuis l'inclusion : -6,0 (0,22) contre -5,7 (0,23). L'amélioration du
score de démangeaison 5-D a été maintenue dans les deux groupes de
traitement tout au long du traitement de 52 semaines.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec la difélikéfaline dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement du prurit associé à la maladie rénale chronique (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Chez les patients atteints de maladie rénale terminale sous hémodialyse, la clairance corporelle totale de la difélikéfaline est réduite par rapport aux sujets sains et les concentrations plasmatiques diminuent lentement jusqu'à être éliminées pendant la séance de dialyse. En raison de l'élimination de 70 à 80 % de la difélikéfaline pendant la dialyse, la difélikéfaline peut être administrée à la fin de chaque séance d'hémodialyse chez ces patients. Les données disponibles sur la variabilité interindividuelle chez les sujets hémodialysés recevant 0,5 microgramme/kg de difélikéfaline laissent penser que la variabilité de l'ASC (Aire Sous la Courbe) peut dépasser 30 %.
Distribution
La liaison de la difélikéfaline aux protéines plasmatiques est faible à modérée, allant de 24 à 32 %, et n'est pas affectée par la maladie rénale. Le volume moyen de distribution à l'état d'équilibre varie de 145 à 189 mL/kg chez les sujets sains et de 214 à 301 mL/kg chez les patients hémodialysés présentant un prurit modéré à sévère. Le passage de la difélikéfaline dans le système nerveux central est limitée (inférieure à la limite de quantification) comme le montrent les données physico-chimiques, in vitro et animales.
Élimination
Chez les sujets sains, la principale voie d'élimination de la difélikéfaline est rénale, représentant environ 81 % de la dose excrétée dans les urines contre 11 % par excrétion fécale. Chez les volontaires sains et les sujets sous hémodialyse, la plus grande partie de la dose excrétée dans les urines et les fèces était de la difélikéfaline non métabolisée avec des quantités mineures de métabolites putatifs, aucune ne dépassant 2,5 %. La clairance totale moyenne a varié de 54 à 71 mL/h/kg et la demi-vie moyenne de 2 à 3 heures. En revanche, chez les insuffisants rénaux hémodialysés, l'élimination s'est faite principalement par les fèces, représentant en moyenne environ 59 % de la dose ; environ 19 % ont été récupérés dans le dialysat et environ 11 % dans les urines. Par rapport aux sujets ayant une fonction rénale normale, la clairance totale moyenne a diminué et les demies-vies ont été multipliées par 10 environ, avec des fourchettes de 5,3 à 7,5 mL/h/kg et de 23 à 31 heures, respectivement.
Interaction avec d'autres médicaments
La difélikéfaline n'est ni un substrat des CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4, ni un inhibiteur des CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4/5 et son potentiel d'induction des CYP1A2, CYP2B6 ou CYP3A humains est minime ou nul. Elle n'est pas non plus un inhibiteur des enzymes de glucuronidation (UGT1A3, UGT1A9 ou UGT2B7).
De plus, la difélikéfaline n'est pas un inhibiteur de
BCRP, BSEP, LAT1, MATE1, MATE2-K, MRP2, OAT1, OAT3, OATP1A2, OATP1B1,
OATP1B3, OCT1, OCT2, OCT3, glycoprotéine P, PEPT1 ou PEPT2, et n'est
pas un substrat pour ASBT, BCRP, BSEP, LAT1, MATE1, MATE2-K, MRP2,
OAT1, OAT2, OAT3, OATP1A2, OATP1B1, OATP1B3, OATP2B1, OCT1, OCT2, OCT3,
OCTN1, OCTN2,OSTαβ, glycoprotéine P, PEPT1 ou PEPT2.
Linéarité/non-linéarité
Il a été démontré que la pharmacocinétique de la difélikéfaline était linéaire et dose dépendante chez les sujets sains (testée sur des intervalles de doses de 1 à 40 et de 1 à 20 microgrammes/kg dans des études à dose unique et répétée, respectivement). La proportionnalité de la dose à l'état d'équilibre a également été établie chez des patients atteints de maladie rénale chronique sous hémodialyse recevant des doses répétées de 0,5 à 2,5 microgrammes/kg, 3 fois par semaine pendant 1 semaine. Toutefois, dans une autre étude, la proportionnalité de la dose a été observée aux doses de 0,5 et 1 microgramme/kg, mais pas à la dose de 1,5 microgramme/kg. Les valeurs de concentration plasmatique minimale ont atteint l'état d'équilibre dès la deuxième dose et, pour la dose de 0,5 microgramme/kg, le rapport d'accumulation moyen était de 1,144 dans une étude basée sur l'ASC0-48h et de 1,33 dans une autre étude, basée sur l'ASC0-44h ; indiquant que la variabilité pour les paramètres d'accumulation peut dépasser 30 %.
Caractéristiques dans des groupes spécifiques de sujets ou de patients
Sur la base des données disponibles, rien n'indique que des facteurs tels que l'âge, le sexe, l'origine ethnique ou l'insuffisance hépatique légère à modérée aient un impact sur la pharmacocinétique de la difélikéfaline.
Kapruvia a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Des cas de somnolence et/ou de sensations de vertige ont été rapportés chez des patients traités par la difélikéfaline (voir rubrique Effets indésirables). Les patients doivent être avertis de ne pas conduire ou utiliser des machines dangereuses jusqu'à ce que l'effet de la difélikéfaline sur l'aptitude du patient à conduire ou à utiliser des machines soit connu. Une somnolence est survenue au cours des 3 premières semaines de traitement et a eu tendance à s'atténuer avec la poursuite du traitement. Des sensations de vertige sont survenues au cours des 9 premières semaines de traitement et se sont révélées généralement transitoires.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, de toxicité à doses répétées, de génotoxicité et de potentiel carcinogène ne révèlent aucun danger particulier pour l'Homme.
Toxicité sur la reproduction
Chez les rats, la fertilité masculine et féminine, le développement embryonnaire précoce et le développement prénatal et post-natal n'ont pas été affectés jusqu'à 2 000 fois l'ASC humaine. Chez le lapin, le développement prénatal n'a pas non plus été perturbé malgré une toxicité maternelle marquée à une exposition 30 fois supérieure à l'ASC humaine.
La difélikéfaline traverse le placenta chez le rat.
Potentiel d'abus et de dépendance
Les études sur le potentiel d'abus et de dépendance chez le rat suggèrent que la difélikéfaline n'est pas susceptible de présenter un risque de potentiel d'abus ou de dépendance physique.
À usage unique seulement.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament soumis à une prescription hospitalière.
Prescription réservée aux spécialistes en néphrologie
Solution injectable.
Solution limpide, incolore, exempte de particules (pH 4,5).
Kapruvia est fourni dans un flacon en verre à usage unique de 2 mL (type I), muni d'un bouchon en caoutchouc bromobutyl, un joint en aluminium et d'un capuchon en plastique amovible bleu, sous forme de solution de 1 mL contenant 50 microgrammes de difélikéfaline.
Boîte de 12 flacons contenant 1 mL de solution injectable.
Chaque flacon de 1 mL contient 50 microgrammes de difélikéfaline (sous forme d'acétate).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Acide acétique (pour ajustement du pH)
Acétate de sodium trihydraté (pour ajustement du pH)
Chlorure de sodium
Eau pour préparations injectables